產品的生產工藝及關鍵設施、設備應按驗證方案進行驗證。當影響產品質量的主要因素,如工藝、質量控制方法、主要原輔料、主要生產設備等發生改變時,以及生產一定周期后,應進行再驗證。藥品生產過程的驗證必須包括空氣凈化系統,制藥企業應自行對潔凈室環境驗證的周期制定一個管理規程。

由ISO14644?中可知,在無菌藥品潔凈室確認的確認時,主要包括: 已安裝的過濾器系統的檢漏和完整性測試、氣流測試(流速、風量) 及換氣次數、自凈及恢復時間測試、壓差測試、氣流方向測試和氣流流型研究、溫度測定測試、相對濕度測試、懸浮粒子測定、微生物污染監測(浮游菌、沉降菌和表面微生物) 等。潔凈室的確認目的是評估潔凈室的潔凈級別是否滿足其預期用途的過程,通常會結合生產需求進行風險評估,測試時需考慮“靜態”測試及“動態”測試。在實際測試過程中,設計確認應當證明設計符合用戶需求,并有相應的文件。安裝和運行確認完成并符合要求后,方可進行性能確認。
根據歐盟附錄一4.23用于無菌產品生產的潔凈室和潔凈空氣設備,如單向流單元(UDAFs) 、RABS和隔離器,應根據所要求的環境特性進行確認。每個生產操作要求具有合適的動態下環境潔凈水平,以最大程度降低所處理的產品或物料的污染風險。應維持“靜態”和“動態”下的適當潔凈度水平。可知現階段對無菌環境的要求更加的貼合實際情況來驗證,要求相較更加嚴格,覆蓋更加的廣泛,故而在實際操作中的動作影響應多加進行風險評估。

一般說來,空氣凈化系統在新建、改建之后必須全面驗證;正常運行后,應做日常的監測記錄工作,如房間的溫濕度、風壓,以及定期檢查微生物和微粒。

空調凈化系統中的空氣平衡工作是一項技術性較強、調試復雜的工作;一經調整,平時不可隨意變動風閥位置,若發現風壓流向不對,應找出原因后,才能調整風閥,以免破壞空氣的平衡,尤其是無菌生產區域,房間多、潔凈級別不同,風壓差逐步降低,任何風閥位置的變動,都會引起各房間風壓的連鎖反應。空調凈化系統調試完畢后,應定期檢查風量,并計算出各房間的換氣次數。風量的檢查可每年1~2次。根據積累的驗證參數,科學地合理地確定一個環境驗證周期。這是確定潔凈室環境驗證周期的原則。

GMP對無菌藥品生產環境要求較嚴,除空調凈化系統安裝結束做驗證外,每年還要定期測試一些項目:

2)高效過濾器調換或修理后,必須做DOP泄漏試驗;
3)空調凈化系統的風量每年檢查1~2次,并核算出各房間的換氣次數;
4)對于潔凈級別百級到C級的房間,在無菌產品生產期間,每天應測定懸浮粒子數,不過采樣量及采樣數目可以按評估減少;
5)浮游菌或沉降菌在無菌產品生產期間,每天應測定,但采樣量及采樣數目可以按評估減少;
6)表面污染測試在無菌藥品生產期間每天應進行;
7)無菌藥品在停止生產、空調凈化系統關閉后,要恢復生產,需按驗證要求重新進行懸浮粒子、浮游菌或沉降菌的測試。

下表給出無菌生產區域日常監測的內容。制藥企業可結合企業實際、結合GMP要求實施:
| 無菌生產區域日常監測內容表 | ||||
| 項目 | 潔凈度 | 浮游菌 | 表面細菌污染 | 人體細菌污染 |
| 頻率 | 百級(每個層流罩下) | 每班1個樣品 | 每班3個樣品 | 每班從任一操作工身上取樣 |
| C級(房間) | 每班1個樣品 | 每個房間每個周期3個樣品 | 輪流取樣 | |
| 位置 | 百級 | 關鍵操作工藝口處 | 任意取樣 | 任意取樣 |
| C級 | 工作面處 | 墻、天花板及非接觸藥粉的設備處任意取樣 | 從在該潔凈區域工作的操作工中取樣 | |
| 采樣方法 | —— | 浮游菌采集器 | 培養皿或棉球擦抹法 | 培養皿或棉球擦抹法 |
| —— | 至少25cm2 | 手套2個手指表面及25cm2外表面 | ||
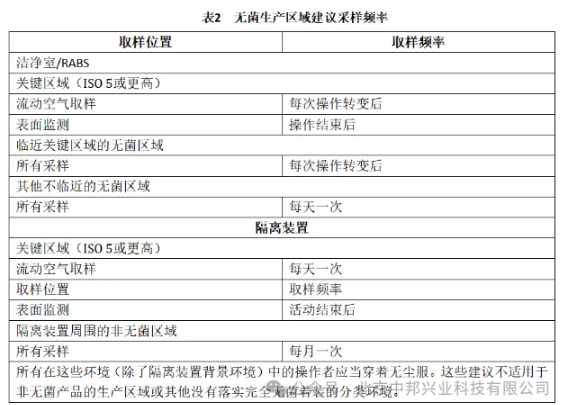
如果您有潔凈室潔凈度確認方面的問題,或者潔凈環境驗證類的儀器設備需求,可以直接找北京中邦興業,專業技術工程師隨時可以幫您解答介紹。










 400-6872-158
400-6872-158 點擊咨詢
點擊咨詢 





